Have you ever wondered how scientists study which genes are active in cells and how that activity changes under different conditions? Bulk RNA-Seq is a powerful technique that helps answer these questions. By analyzing RNA, this method reveals the inner workings of cells, allowing researchers to explore everything from disease mechanisms to how plants respond to stress.
This guide will walk you through the basics of Bulk RNA-Seq, including how it works, the steps involved in analysis, and its wide-ranging applications in science and medicine. Whether you’re a student, researcher, or curious learner, you’ll discover how this technology is transforming our understanding of biology.
Read more about Transcriptome Analysis
Sequencing Technologies: Foundations of Bulk RNA-Seq
To understand Bulk RNA-Seq, it’s essential to explore the sequencing technologies that make it possible. Over the years, advancements in sequencing have transformed how we study RNA, improving the accuracy, speed, and depth of analysis. From early methods to modern high-throughput systems, these technologies form the backbone of RNA-Seq.
The Evolution of Sequencing Technologies
Sanger Sequencing
Sanger sequencing, developed in the 1970s, was the first method to read DNA and RNA sequences. This technique uses chain-terminating nucleotides to determine the sequence of DNA fragments. While it provided high accuracy, Sanger sequencing was limited by its low throughput and slow processing. It laid the groundwork for future advancements, but its scalability was insufficient for transcriptome-wide studies.
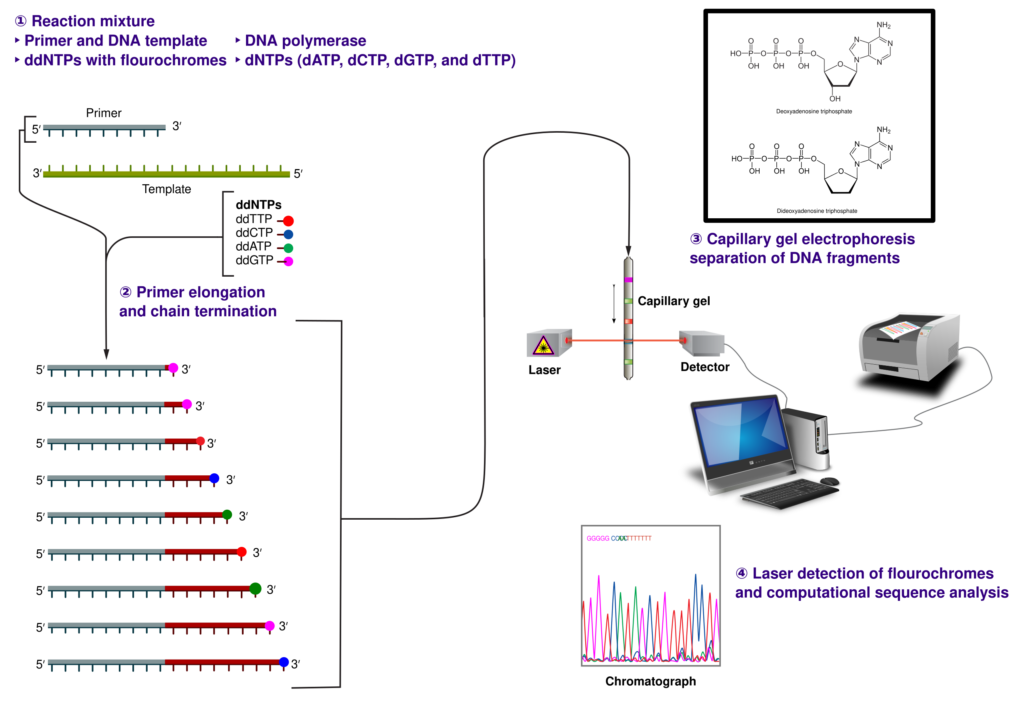
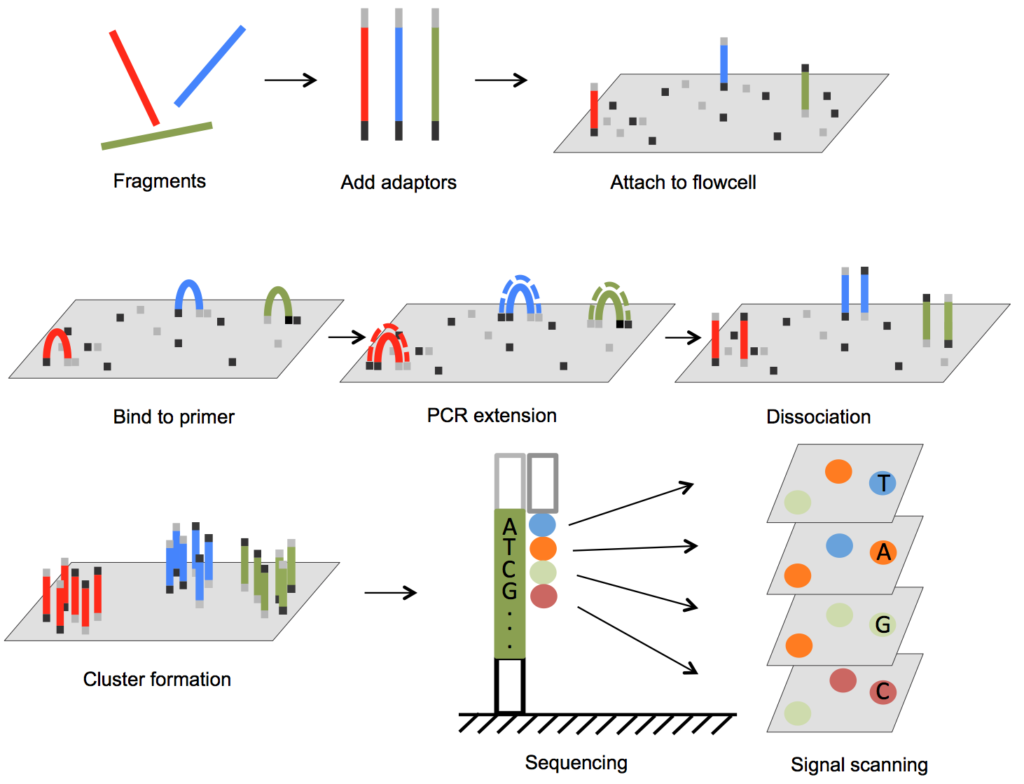
Next-Generation Sequencing (NGS)
NGS revolutionized RNA-Seq by allowing millions of RNA fragments to be sequenced simultaneously. Platforms like Illumina provide high-throughput capabilities, enabling researchers to generate comprehensive transcriptomic datasets in a single experiment. The speed, accuracy, and scalability of NGS make it the standard for Bulk RNA-Seq today.
NGS workflows involve fragmenting RNA, converting it to complementary DNA (cDNA), and sequencing these fragments.
Its sensitivity allows for the detection of low-abundance transcripts and the discovery of novel RNA species.
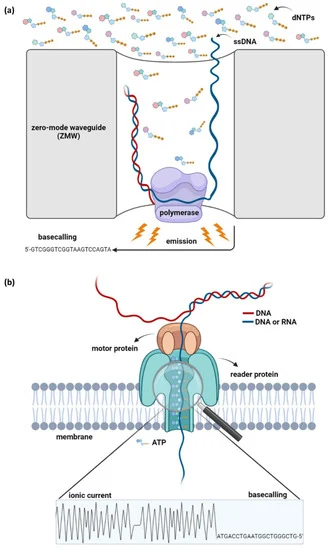
Third-Generation Sequencing
TGS technologies, such as PacBio and Nanopore sequencing, take RNA analysis a step further by reading long fragments. This is particularly useful for identifying complex RNA structures and alternative splicing events. However, TGS comes with higher costs and lower accuracy compared to NGS, making it less common for Bulk RNA-Seq.
The Role of NGS in Bulk RNA-Seq
NGS is the cornerstone of Bulk RNA-Seq, providing the tools to capture and analyze RNA across entire cell populations. Its high accuracy and scalability enable researchers to explore gene expression with precision and depth.
- Massive Parallel Processing: NGS can sequence millions of RNA fragments simultaneously, generating large datasets for detailed analysis.
- Versatility: It is effective across a range of sample types, from animal tissues to plant cells, making it adaptable for diverse research needs.
- Cost-Effectiveness: While still expensive, the cost of NGS has decreased significantly, making it accessible for many labs.
How Sequencing Technologies Shape Transcriptomics
The evolution from Sanger to NGS and TGS technologies has dramatically enhanced our ability to study RNA. NGS remains the preferred choice for Bulk RNA-Seq due to its balance of accuracy, cost, and scalability, while TGS offers exciting opportunities for specialized applications. Understanding these technologies equips you with the knowledge to choose the best approach for your research.
The Bulk RNA-Seq Workflow: From Bench to Analysis
Experimental Design and Sample Preparation
Your Bulk RNA-Seq experiment starts with careful planning. Clearly define your objectives and select the right sample groups, such as comparing healthy and diseased tissues or studying treated versus untreated cells. By including proper controls and replicates, you ensure your results are both reliable and interpretable.
Once you’ve planned your experiment, you’ll need to extract RNA from your samples. Use methods like column-based kits or TRIzol reagents to isolate RNA while preserving its quality. It’s crucial to prevent contamination and degradation during this step, as RNA is highly fragile. After extraction, assess RNA quality using tools like a Bioanalyzer or Nanodrop. Aim for high RNA integrity numbers (RIN) to ensure your samples are suitable for sequencing.
Sequencing Library Preparation
After you’ve isolated high-quality RNA, the next step is to prepare it for sequencing. This involves converting RNA into complementary DNA (cDNA), which is more stable and compatible with sequencing platforms.
Here’s what the library preparation process looks like:
- Reverse Transcription: Use enzymes to create cDNA from RNA, preserving the original transcript information.
- Fragmentation: Break the cDNA into smaller pieces, making them easier to sequence.
- Adapter Ligation: Add short DNA sequences (adapters) to both ends of the fragments. These adapters help the fragments bind to the sequencing platform and allow identification during analysis.
- Amplification: Use PCR to increase the quantity of cDNA, ensuring enough material for high-throughput sequencing.
Consistency in these steps is critical. Small errors during library preparation can lead to biased or incomplete results, so follow protocols carefully.
Sequencing and Data Generation
With the libraries prepared, you can load them onto a sequencing platform, such as Illumina. These platforms read the cDNA fragments and generate raw data as millions of short reads. Sequencing depth—the number of reads per sample—is an important factor to consider. Deeper sequencing provides more information, especially for detecting low-abundance transcripts or rare RNA species.

The output of this stage is typically in the form of FASTQ files. These files contain both the RNA sequences and quality scores, which are essential for the next step: bioinformatics analysis.
Bioinformatics Analysis of Bulk RNA-Seq Data
Once you’ve sequenced your RNA samples, the raw data needs to be processed and analyzed to extract meaningful insights. This process, often called in silico analysis, involves a series of steps, each playing a critical role in understanding gene expression patterns. Let’s break down the workflow into clear, actionable steps.
Quality Control
Your first step is to check the quality of the raw sequencing data. This ensures you’re working with reliable information from the start. Tools like FastQC allow you to evaluate read quality, detect adapter contamination, and spot overrepresented sequences. Poor-quality data can lead to misleading results, so take the time to clean it up.
After running quality checks, you’ll likely find some issues, such as low-quality bases at the ends of reads. Use tools like Trimmomatic or Cutadapt to trim these problematic sequences and remove contaminants. By cleaning your data, you’re setting the stage for accurate downstream analysis.
Read Mapping and Alignment
Next, you’ll map your cleaned reads to a reference genome or transcriptome. This step identifies where each RNA fragment originates, linking sequences to specific genes. Software like HISAT2, STAR, or Bowtie can help you perform this alignment efficiently.
If you’re studying an organism without a reference genome, you can use de novo assembly tools like Trinity to reconstruct the transcriptome from scratch. Accurate alignment is essential because errors at this stage can cascade into the rest of your analysis, so double-check your settings and results.
Gene Expression Quantification
After mapping, you’ll measure the number of reads that align to each gene. This step quantifies how much RNA each gene produces under your experimental conditions. Tools like featureCounts or HTSeq count these reads and generate a gene expression matrix.
This matrix acts as the foundation for all subsequent analyses. It shows you which genes are active, and to what extent, in your samples. This information is crucial for understanding cellular responses or identifying key pathways in your study.
Normalization
Raw read counts need to be normalized to account for differences in sequencing depth or library size between samples. Without normalization, comparisons across samples could be misleading.
Common normalization methods include:
- TPM (Transcripts Per Million): Adjusts for gene length and sequencing depth.
- RPKM (Reads Per Kilobase of transcript per Million mapped reads): Similar to TPM but includes additional scaling.
- DESeq2 Normalization: A statistical method that adjusts counts for meaningful comparisons.
Normalization ensures you’re comparing apples to apples when looking at gene expression differences.
Differential Expression Analysis
This step allows you to identify genes that show significant differences in expression between conditions. For example, you can compare treated vs. untreated samples or healthy vs. diseased tissues to find genes that may play a role in your research question.
Tools like DESeq2 or EdgeR are widely used for this analysis. They apply statistical models to your data, accounting for variability and ensuring your results are robust. Differentially expressed genes often point to pathways or mechanisms that warrant further investigation.
Functional Enrichment Analysis
Once you’ve identified differentially expressed genes, you can dive deeper into their biological significance. Functional enrichment analysis helps you understand the pathways or processes these genes are involved in.
Use tools like DAVID or GSEA (Gene Set Enrichment Analysis) to analyze your gene list. For example:
- Find out if genes in your study are overrepresented in pathways like metabolism, immune response, or cell cycle regulation.
- Identify gene ontology terms associated with your data, such as molecular functions or cellular components.
This step helps you move beyond individual genes to see the bigger biological picture.
Data Visualization
Visualizing your data makes it easier to interpret and share your findings. Common visualization methods include:
- Heatmaps: Show patterns of gene expression across samples. Use them to identify clusters of co-expressed genes.
- Volcano Plots: Highlight differentially expressed genes based on statistical significance and expression level.
- Principal Component Analysis (PCA): Summarize variability in your dataset, revealing how samples group together based on gene expression.
By presenting your data visually, you make complex results accessible to both experts and non-specialists.
Let Us Help You Unlock the Power of RNA-Seq
Ready to turn your RNA-Seq data into actionable insights? On our platform, you can easily navigate the complexities of bioinformatics analysis. Whether you’re new to RNA-Seq or need expert guidance, we’re here to help.
Share your project goals with us, and we’ll handle the heavy lifting—from quality control to advanced data interpretation. With our expertise and tools, you’ll get accurate, meaningful results tailored to your research needs. Let’s work together to make your RNA-Seq project a success!
Applications of Bulk RNA-Seq
Bulk RNA-Seq is a versatile tool that helps researchers uncover valuable insights into gene expression and cellular behavior. By analyzing RNA from groups of cells, you can explore a wide range of biological questions across various fields. Below are some of the key applications of Bulk RNA-Seq, with examples to show how it transforms research and discovery.
1. Disease Research: Unveiling Molecular Mechanisms
Bulk RNA-Seq plays a crucial role in studying diseases, as it allows you to identify changes in gene expression between healthy and diseased states. For example, by analyzing RNA from cancer tissues, you can discover which genes are driving tumor growth or resistance to therapies. Similarly, in neurodegenerative diseases like Alzheimer’s, RNA-Seq helps you understand how disrupted gene activity contributes to the disease process.
Moreover, this technology enables you to study infectious diseases by examining how pathogens alter host gene expression. This insight can lead to the development of vaccines or targeted treatments.
2. Drug Development and Personalized Medicine
Another important application of Bulk RNA-Seq is its ability to accelerate drug development. By analyzing how cells respond to different drugs, you can identify new therapeutic targets or understand why certain treatments fail. Furthermore, this technology supports personalized medicine by uncovering individual gene expression profiles.
For instance, in cancer treatment, you can use RNA-Seq data to predict which therapies will be most effective for a patient based on the unique characteristics of their tumor. As a result, Bulk RNA-Seq makes it easier to design customized treatments that improve outcomes.
3. Developmental Biology: Tracking Gene Expression Over Time
Bulk RNA-Seq allows you to study how gene activity changes during development. For example, you can use it to examine which genes are turned on or off at different stages of embryonic growth. This helps you understand the molecular programs that guide cell differentiation and tissue formation.
In addition, developmental biologists often rely on RNA-Seq to identify genes involved in lineage specification, providing deeper insights into how organisms grow and develop. These findings are critical for advancing fields like regenerative medicine.
4. Environmental and Ecological Research
In ecological studies, Bulk RNA-Seq provides a powerful way to explore how organisms respond to their environment. For example, you can use it to study how plants adapt to drought, extreme temperatures, or soil contaminants. Similarly, you can analyze the gene expression profiles of animals exposed to pollutants or stressors.
Importantly, this technology also aids in understanding ecosystem dynamics. By studying microbial communities in different environments, you can learn how changes in gene expression affect processes like nutrient cycling or carbon sequestration.
5. Agricultural Advancements: Improving Crops and Livestock
In agriculture, Bulk RNA-Seq is widely used to enhance crop and livestock productivity. For crops, you can study how gene expression changes in response to pests, pathogens, or nutrient deficiencies. This helps you identify genes that improve stress tolerance or increase yield.
For livestock, RNA-Seq enables you to uncover the genetic basis of traits like disease resistance or growth efficiency. These insights allow you to optimize breeding programs and develop strategies to improve animal health and performance.
Challenges and Future Directions in Bulk RNA-Seq
Bulk RNA-Seq has transformed the way we study gene expression, but it is not without its challenges. As researchers continue to push the boundaries of this technology, it’s important to understand both the limitations it currently faces and the promising directions it may take in the future. Addressing these challenges will ensure that RNA-Seq remains a valuable tool in advancing biology and medicine.
Challenges
Data Management and Analysis Complexity
One of the biggest challenges in Bulk RNA-Seq is managing the enormous amount of data it generates. Each sequencing run produces millions of reads, which must be stored, processed, and analyzed. This requires powerful computational resources and specialized bioinformatics expertise, which may not be readily available in all labs. As a result, smaller research groups often struggle to keep up with the demands of RNA-Seq analysis.
Moreover, interpreting the data can be equally complex. Because gene expression patterns vary across experimental conditions, choosing the right statistical models is essential. Without careful planning and proper tools, results can be unreliable or difficult to replicate.
Reproducibility Issues
Reproducibility is another concern in Bulk RNA-Seq studies. Variations in sample preparation, library protocols, or bioinformatics workflows can lead to inconsistent results across experiments. Even minor differences in how data is processed can significantly impact the interpretation of gene expression patterns. Consequently, standardization is crucial for ensuring reproducible and comparable results across studies.
Limitations of Bulk RNA-Seq
Because Bulk RNA-Seq averages gene expression across all cells in a sample, it lacks the resolution to detect differences between individual cells. This can obscure important details, such as rare cell populations or cell-to-cell variability. Although single-cell RNA-Seq addresses this limitation, it comes with its own challenges, including higher costs and more complex workflows.
Future Directions
Improved Computational Tools
To address the challenges of data management and complexity, researchers are developing more efficient bioinformatics tools and pipelines. For example, machine learning algorithms are increasingly being used to automate data analysis, identify patterns, and make predictions. These advancements will make RNA-Seq analysis faster and more accessible, even for researchers without extensive computational expertise.
Integration with Multi-Omics
The future of Bulk RNA-Seq lies in its integration with other omics technologies, such as proteomics and metabolomics. By combining RNA-Seq data with information about proteins or metabolites, researchers can gain a more comprehensive understanding of cellular processes. This holistic approach will be especially valuable for studying complex diseases, where multiple layers of regulation interact.
Affordable and Portable Sequencing
Efforts to reduce the cost of RNA-Seq are ongoing. Advances in sequencing platforms, including portable devices like nanopore sequencers, promise to make RNA-Seq more affordable and accessible. These portable tools will enable researchers to perform transcriptomics analysis in field settings, such as remote ecological sites or clinical environments.
Single-Cell and Spatial Transcriptomics Integration
While Bulk RNA-Seq provides valuable insights at the population level, the integration of single-cell and spatial transcriptomics will complement its capabilities. Single-cell RNA-Seq can reveal the unique gene expression profiles of individual cells, while spatial transcriptomics adds a layer of spatial context by mapping gene expression to specific regions within tissues. Together, these approaches will provide a more detailed understanding of cellular behavior.
Standardization of Protocols
To improve reproducibility, the field is moving toward the standardization of RNA-Seq protocols and bioinformatics workflows. Efforts to create universally accepted guidelines will ensure that results are comparable across studies and labs, fostering collaboration and reliability in transcriptomics research.
FAQs About Bulk RNA-Seq
1. What is Bulk RNA-Seq, and how does it work?
Bulk RNA-Seq is a technique used to analyze RNA from a group of cells, revealing which genes are active and at what levels. The process involves extracting RNA, converting it to complementary DNA (cDNA), sequencing the cDNA, and analyzing the data to understand gene expression patterns.
2. How is Bulk RNA-Seq different from single-cell RNA-Seq?
Bulk RNA-Seq measures the average gene expression across all cells in a sample, making it ideal for studying tissue-level patterns. In contrast, single-cell RNA-Seq analyzes individual cells, providing detailed insights into cell-to-cell variability and rare cell populations.
3. What are the common applications of Bulk RNA-Seq?
Bulk RNA-Seq is widely used in:
- Disease research to identify genes linked to specific conditions.
- Drug development for studying cellular responses to treatments.
- Developmental biology to track gene activity during growth stages.
- Agriculture to improve crop and livestock traits.
- Environmental studies to explore how organisms respond to stressors.
4. What tools are used for Bulk RNA-Seq data analysis?
The analysis involves several key tools, including:
- FastQC for quality control.
- HISAT2 or STAR for read mapping.
- DESeq2 or EdgeR for differential expression analysis.
- GSEA or DAVID for functional enrichment analysis.
5. Is Bulk RNA-Seq expensive, and how can I manage the costs?
Bulk RNA-Seq costs can vary depending on the number of samples and depth of sequencing required. However, costs have decreased significantly over the years. Planning your experiments carefully, pooling samples where possible, and leveraging cost-effective platforms can help manage expenses.
Explore More
Have additional questions or need expert guidance for your RNA-Seq project? Contact us today or explore our platform for detailed insights and professional support. Let’s make your research journey seamless and successful!
Conclusion
Bulk RNA-Seq has become a cornerstone technology for exploring gene expression and uncovering the molecular mechanisms that drive biology. From advancing disease research to improving crops and livestock, its applications span across diverse fields, transforming the way we study life at the molecular level.
Despite challenges like data complexity and costs, ongoing advancements in computational tools, sequencing platforms, and integrative techniques are making RNA-Seq more accessible and powerful than ever. By addressing these limitations and adopting complementary technologies like single-cell and spatial transcriptomics, researchers can gain even deeper insights into cellular behavior.
Reference:
- Yuan L, Yingjia S, Wesley W, Ronald W. Next Generation Sequencing in Aquatic Models. In: Jerzy KK, editor. Next Generation Sequencing. Rijeka: IntechOpen; 2016. p. Ch. 2.
- Athanasopoulou K, Michaela A. Boti, Panagiotis G. Adamopoulos, Paraskevi C. Skourou, and Andreas Scorilas. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life. 2022;12.